Ankang Wang, People's Republic of China
General Surgery Department, First Hospital of Lanzhou University
Lin28A drives hepatic fibrosis by suppressing HMGA2/HIF-1α-dependent ferroptosis in hepatic stellate cells
Ankang Wang1, Jia Yao1, Hongfa Sun1, Haixia Zhao1, Ye Xie1, Mengchao Yan1, Yue Zhang1, Xun Li1.
1Key Laboratory of Biotherapy and Regenerative Medicine of Gansu Provinc, the First Hospital of Lanzhou University, lanzhou, People's Republic of China
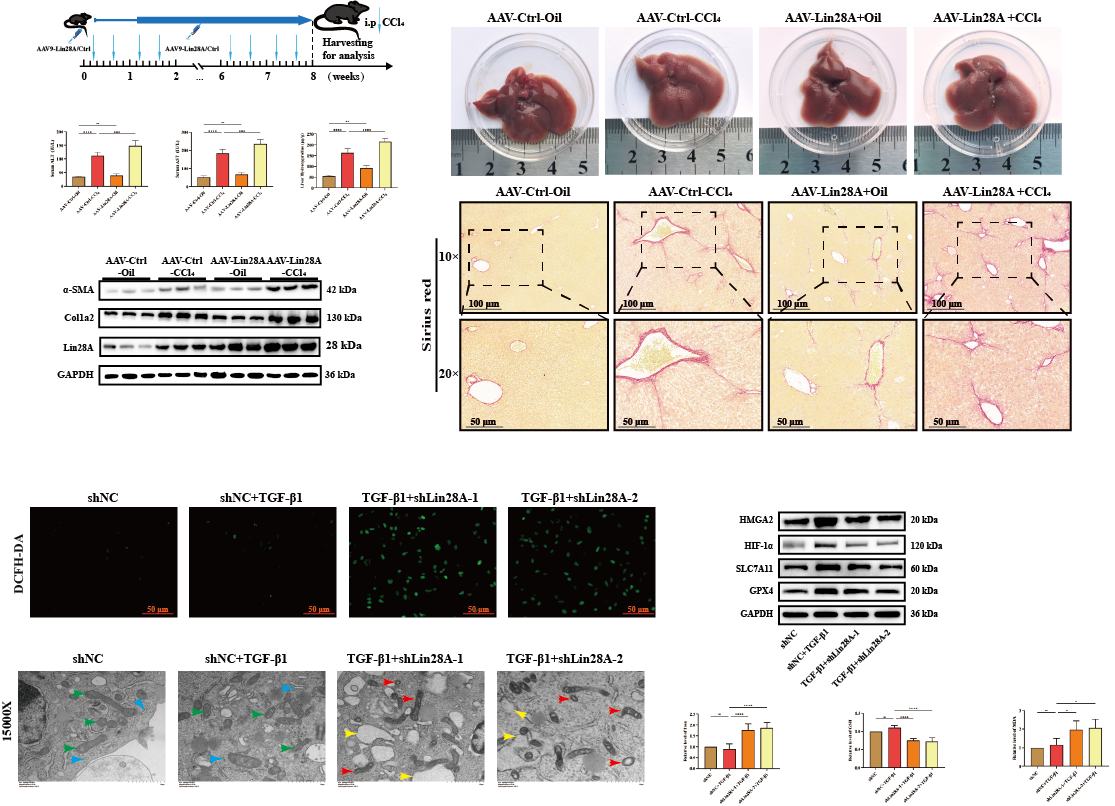
Background & Aims: Liver fibrosis lacks effective therapies, and the role of the RNA-binding protein Lin28A in hepatic stellate cells (HSCs) fate remains unexplored. We investigated Lin28A’s function in fibrogenesis and its therapeutic targeting via ferroptosis regulation.
Methods: First, clinical specimens were collected to analyze Lin28A expression levels and subcellular localization. Subsequently, Lin28A was overexpressed via tail vein injection of AAV9-Lrat vectors in a CCl₄-induced liver fibrosis model to assess its impact on fibrogenesis. For in vitro experiments, lentiviral shRNA was employed to knock down Lin28A expression in LX-2 cells, and its effects on hepatic stellate cells (HSCs) activation status and ferroptosis were examined. To further elucidate the underlying mechanism, HMGA2 rescue experiments were performed. Finally, the effects of C1632, a specific small-molecule inhibitor of Lin28A, on HSCs activation and the progression of liver fibrosis in mice were investigated using integrated in vitro and in vivo models.
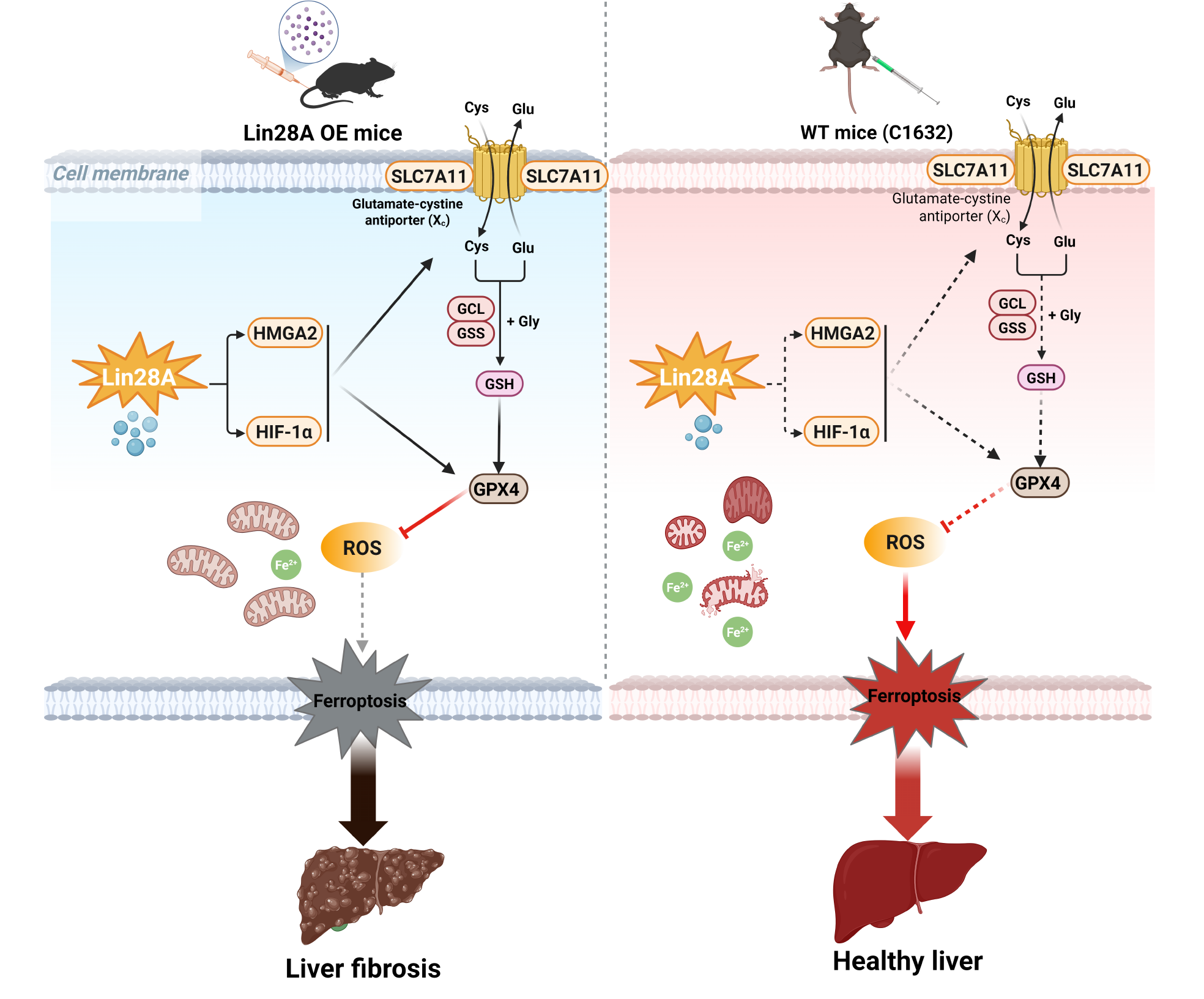
Results: Lin28A expression was significantly upregulated in human cirrhotic liver tissues and mouse fibrotic livers, where it co-localized with activated α-SMA⁺ hepatic stellate cells (HSCs). HSC-specific overexpression of Lin28A exacerbated CCl₄-induced liver fibrosis, manifested by increased collagen deposition, elevated serum ALT/AST levels, and upregulated α-SMA and Col1a2 expression. Conversely, Lin28A knockdown suppressed TGF-β1-induced HSC activation, reducing cell migration, proliferation, and α-SMA/Col1a2 expression, and concurrently triggered ferroptosis. This Lin28A deficiency-induced ferroptosis was confirmed by characteristic hallmarks, including mitochondrial condensation, elevated reactive oxygen species (ROS), iron accumulation, glutathione (GSH) depletion, and downregulation of GPX4 and SLC7A11. Furthermore, HMGA2 expression exhibited a positive correlation with Lin28A levels both in vitro and in vivo. Mechanistically, rescue experiments demonstrated that HMGA2 overexpression partially reversed the enhanced ferroptosis sensitivity and restored migration capacity induced by Lin28A knockdown. Critically, the Lin28A-specific inhibitor C1632 promoted ferroptosis in activated HSCs in vitro and attenuated CCl₄-induced fibrosis in vivo, reducing collagen accumulation, α-SMA expression, and serum ALT/AST levels.
Conclusions: Downregulation of Lin28A attenuates hepatic fibrosis by promoting HMGA2/HIF-1α-dependent ferroptosis in HSCs, establishing Lin28A as a therapeutic target.
References:
[1] Lin28A
[2] Hepatic fibrosis
[3] Ferroptosis
[4] Hepatic stellate cells (HSCs)
[5] HMGA2/HIF-1α axis
[6] C1632
Lectures by Ankang Wang
| When | Session | Talk Title | Room |
|---|---|---|---|
|
Thu-23 18:30 - 19:30 |
Poster Session | Lin28A drives hepatic fibrosis by suppressing HMGA2/HIF-1α-dependent ferroptosis in hepatic stellate cells | Hall A2-A4 |